
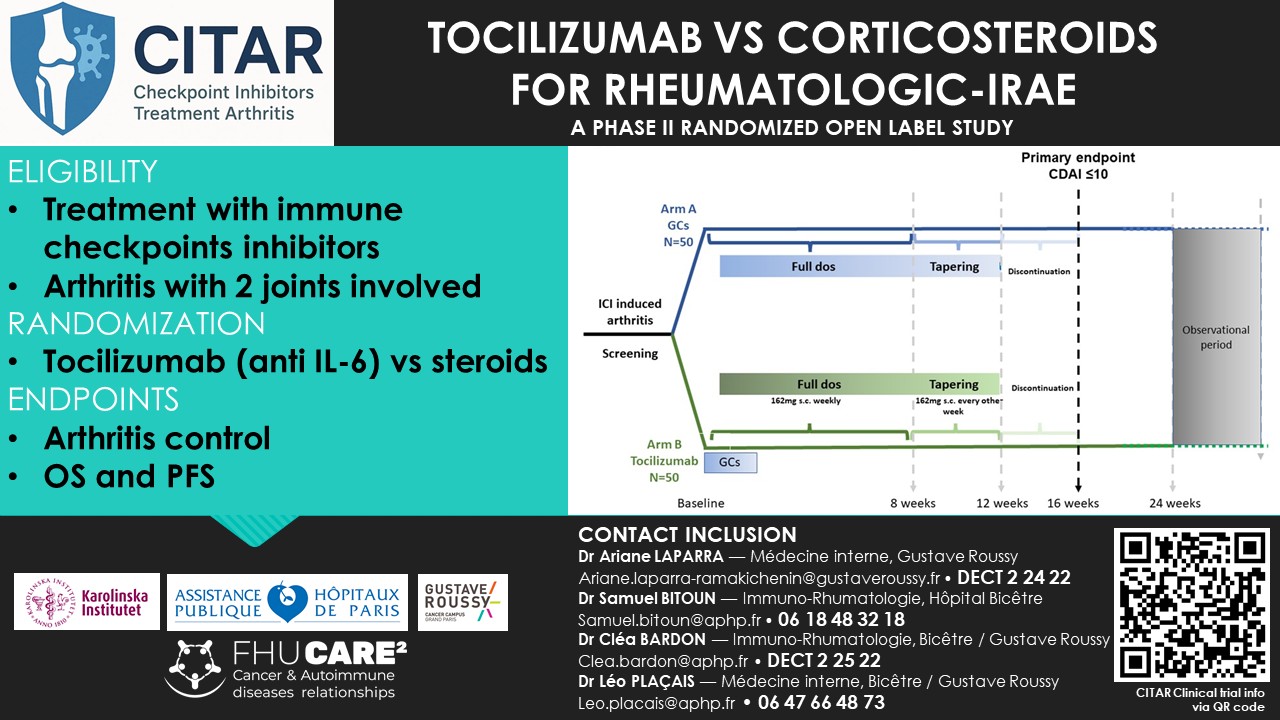
TOCILIZUMAB VS CORTICOSTEROIDS
FOR RHEUMATOLOGIC-IRAE
A PHASE II RANDOMIZED OPEN LABEL STUDY
 |
CITAR trial information : Checkpoint Inhibitor Treatment of ARthritis
Title : CITAR – Comparison of the efficacy and safety of early use of IL-6R blockade with tocilizumab in combination with short-term glucocorticoids versus glucocorticoids alone for the treatment of arthritis induced by cancer immunotherapy by check point inhibitors: a randomized, open, multicentre, proof of concept, superiority, controlled clinical trial.

- CITAR Flyer: CITAR – Tryptique 2025 06 04 v1 [pdf]
- Slides – Oncologist Staff Meeting, Gustave Roussy: CItar-prez_GR_sept_2025.pdf
- Watch Dr. Samuel Bitoun’s conference presentation of the CITAR study :

Medical condition(s) : Patients with histologically (or via cytology) confirmed cancer that develop arthritis secondary to treatment with immune checkpoint inhibitors.
Trial Phase: Therapeutic exploratory (Phase II)
Transition trial : No
Sponsor : Karolinska University Hospital
Participants type : Patients
Age range : 65+ years, 18-64 years
Locations : Sweden, France
Main objective : Does early use of IL6R blockade with tocilizumab in combination with short-term glucocorticoids have a superior efficacy in controlling the arthritis (arm B) compared to GCs alone (arm A) (assessed as CDAI low disease activity or remission) at week 16?
More about CITAR Clinical Trial:
https://euclinicaltrials.eu/search-for-clinical-trials/?lang=en&EUCT=2022-501130-33-00
Main contacts:

Sponsor Karolinska Institutet (KI)
Dr Katerina CHATZIDIONYSIOU
Senior consultant Rheumatologist.
Head of the Clinical Trial Department Rheumatology Unit, Karolinska University Hospital
Email : aikaterini.chatzidionysiou@ki.se

Sponsor AP-HP (project code : APHP 240143)
Dr Samuel BITOUN
Coordinating investigator
Immunology and rheumatology department at Bicêtre Hospital
E-mail: samuel.bitoun@aphp.fr
Phone: +33(0)1.45.21.72.87

Dr Clea BARDON
Institut Gustave Roussy
Email: clea.bardon@aphp.fr
Phone: +33(0)1.45.22.25.22
French Centers (30 patients in France) :
Bicêtre AP-HP
- Samuel Bitoun
Department of Immunology and Rheumatology, Bicêtre Hospital, Le Kremlin-Bicêtre, France - Olivier Lambotte
Department of Internal Medicine and Clinical Immunology, Université Paris-Saclay, Bicêtre Hospital, Le Kremlin-Bicêtre, France
Brest
- Divi Cornec
INSERM UMR1227 – B Lymphocytes, Autoimmunity and Immunotherapies Université de Bretagne Occidentale Department of Rheumatology, CHRU Brest, Brest, France
Bordeaux
- Christophe Richez
Department of Rheumatology, Bordeaux University Hospital, Bordeaux, France
Strasbourg
- Jacques-Eric Gottenberg
Department of Rheumatology, CHRU Strasbourg, Strasbourg, France
Study methodology:
- Phase II trial
- Randomised
- Controlled
- Open-label
- Multicentre
- Rheumatology units
Study population and number of patients:
- Patients treated with immune checkpoint inhibitors (ICIs) for cancer who develop inflammatory arthritis induced by these ICIs.
- 112 patients (56 in each group)
- Including 30 in France: Bicêtre, Bordeaux, Brest, Strasbourg.
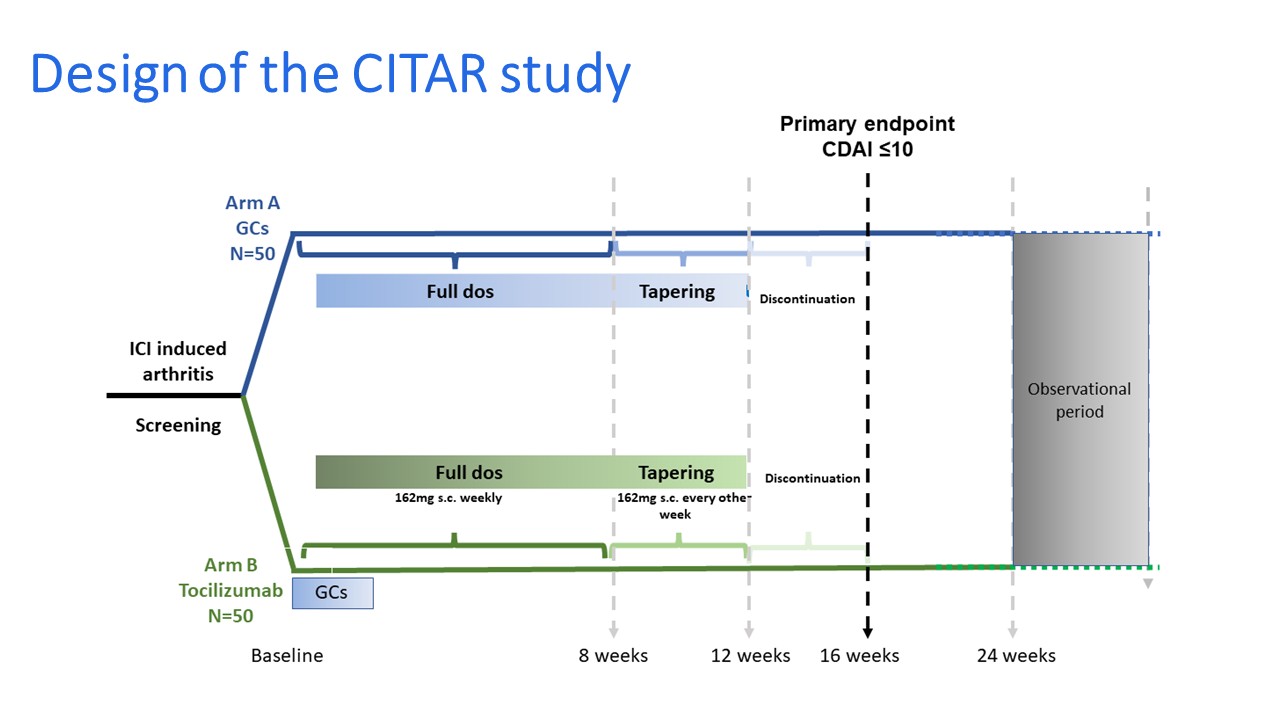
Primary objective :
- Evaluation of the effect of early use of tocilizumab in combination with glucocorticoids (15 days) (arm B)
- VS
- Glucocorticoids alone (arm A) on the CDAI at week 16.
Treatment in the study :
| Arm A : Prednisolone | Arm B : Tocilizumab + Prednisolone reduction |
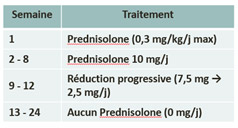 | 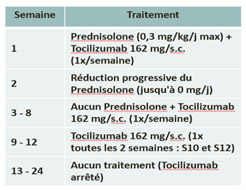 |
Inclusion criteria :
- Patients with histologically (or cytologically) confirmed cancer who develop arthritis (diagnosed by a rheumatologist) secondary to treatment with immune checkpoint inhibitors*
*Patients may be on monotherapy with ICIs or on combination therapy with two ICIs (e.g., ipilimumab and nivolumab). Patients may be receiving ICI in combination with chemotherapy - At least 2 joints involved (clinically defined) + CDAI > 10
- Performance status (ECOG/WHO) 0-1; performance status 2 due to ongoing irAEs is permitted
- Patients already receiving glucocorticoids, regardless of dose, for the treatment of arthritis may be included if the duration of glucocorticoid treatment does not exceed 1 week
- Women of childbearing potential must have a negative pregnancy test (blood or urine) at enrolment
- Women must be post-menopausal for at least 1 year or be willing and able to use highly effective contraception during treatment and for up to 3 months after the last dose of the study drug
Exclusion criteria :
- Mild arthritis requiring no treatment other than NSAIDs or analgesics
- Presence of a life-threatening or organ-threatening treatment-related adverse event (irAE) requiring high doses of glucocorticoids
- Current treatment with glucocorticoids for other indications that cannot be discontinued
- History of inflammatory rheumatic disease prior to cancer diagnosis
- Current participation or participation in an investigational agent study or use of an investigational device within 4 weeks prior to the first dose of treatment
- Diagnosis of immunodeficiency or ongoing systemic immunosuppressive therapy, other than steroids, prior to the first dose of study treatment
- Treatment with a non-biological immunosuppressive or immune system modulating drug
- Treatment with other biological immune system modulating agents (other than ICIs) within 4 weeks prior to the start of treatment
- Vaccination with a live vaccine within 4 weeks prior to the first dose of the study drug or anticipated need for live vaccination during the study, including at least 30 days after the last dose of the study drug
- History of anaphylaxis or immunoglobulin E-mediated hypersensitivity to murine proteins or any component of tocilizumab. History of allergic reactions attributed to compounds with a chemical or biological composition similar to that of tocilizumab.
- History of hypersensitivity to prednisolone or any of its excipients.
- History of HIV or other immunodeficiencies
- History or current evidence of any condition, therapy, or laboratory abnormality that could distort the results of the trial, interfere with the subject’s participation throughout the trial, or that would not be in the subject’s best interest to participate, in the opinion of the treating investigator
- History of psychiatric disorders or substance abuse that would interfere with cooperation with trial requirements
- History of chronic viral hepatitis, alcoholic or metabolic liver disease
- Central nervous system (CNS) metastases
- History of or current positive purified protein derivative (PPD) tuberculin test
- Transplanted organs
- Active infection
- Pregnancy or breastfeeding
- Demyelinating disorders of the central nervous system or pre-existing seizure disorders
- History of diverticulitis, diverticulosis requiring antibiotic treatment, or other symptomatic gastrointestinal (GI) conditions that may promote perforation
- Adequate organ and bone marrow function
- Aspartate aminotransferase (AST) (SGOT)/alanine aminotransferase (ALT) (SGPT) ≤2 × institutional upper limit of normal
- Creatinine clearance Creatinine clearance ≥30 mL/min
Design & Procedure :
Others contacts :
 | |